
Seeing Is Believing: Exposing the “Invisible Monster”
“Our entire civilization has been brought down by a tiny virus,” says Yigong Shi, PhD, a leading Chinese structural biologist. But “we have the tools to see the enemy,” he adds, “and we have the hope that we can beat it.” In the battle between scientists and COVID-19, imaging gives scientists an upper hand.
We know how COVID-19’s central villain, the novel coronavirus known as SARS-CoV-2, forces itself into a cell by becoming one with its membrane barrier. We know how it copies itself. And we know that this virus is promiscuous: Thought initially to be primarily a respiratory virus, it can also infect cells in the heart, the kidneys, the brain, and the gut—illuminating why COVID-19 exhibits so many different symptoms.
We also know in fine detail, what the virus’s killer spike protein looks like and how to gum it up—and, as a result, we also know exactly what a vaccine against it should look like. Just 11 months after this pandemic started its ugly course, we had vaccines that had been shown to protect people from getting COVID-19, as well as antibody-based drugs to treat those who do get it.
Science will help extricate us from this pandemic, but new knowledge doesn’t just fall from the sky. Clever minds are needed to make discoveries, and the right tools speed up that process significantly. The tools of structural biology—really, just practical applications of the basic laws of physics—are integral to building our understanding of the human body and its molecular assailants. Developed over the past 50 years, extremely sensitive microscopes and other imaging techniques have let researchers watch how SARS-CoV-2 forces its way into human cells and usurps the cells’ resources to make more of itself—a thousandfold more—before it sneaks out to infect still more cells.
The “invisible monster”
Sai Li, PhD, was one of the first researchers to visualize the novel coronavirus in cells from infected patients in Wuhan, China. One of Yigong Shi’s protégés, Li is a rising star in structural biology at Beijing’s Tsinghua University. A young scientist, Li had just started his own lab a year before the pandemic broke; at the beginning of 2020, he had just one grad student, who didn’t even have experience with viruses, working in his lab. But even so, like much of the scientific community, Li jumped at the chance to help by learning more about what he calls the “invisible monster.”
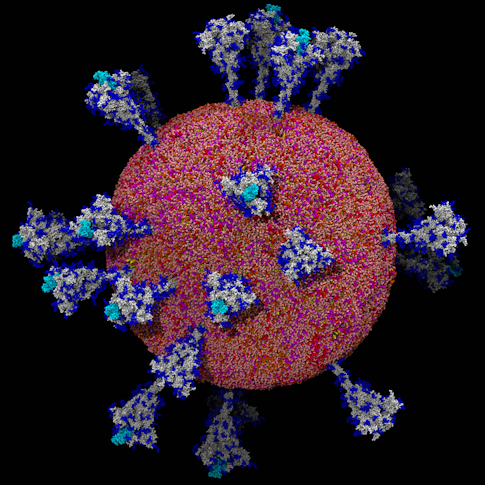
By early February 2020, patient samples arrived in Li’s lab from a Chinese hospital 800 miles southeast of Beijing, in Hangzhou, home of one of the first COVID-19 victims. Li aimed to create a molecular “Most Wanted” picture of this tiny monster using a powerful imaging method called cryo-electron microscopy, or cryo-EM. Adapting this technique, the concept for which had first emerged decades before, in the 1960s, Li imaged more than 2,000 individual coronavirus particles in cells to delineate the three-dimensional architecture of the virus and everything inside it.
By the time Li obtained those first virus samples, the world knew a bit about SARS-CoV-2. Now-standard molecular identification techniques—including genome-sequencing tools as well as long-established procedures like plaque assays to determine how infectious viruses are—had confirmed that this new beast was a coronavirus, and a very infectious one at that. Li’s ambition was to transform a “blob on a slide” into a clear molecular portrait.
Two amino acids change everything
Halfway around the world, in Texas, other researchers were after additional information about SARS-CoV-2 using similar structural biology approaches.
Li and Jason McLellan, PhD, a molecular biochemist at the University of Texas at Austin, work on the same general problems: How do the basic building blocks of life, proteins, run our body’s chemical reactions, as well as make most of the parts of our bodies (like bone, muscle, and nerves)? How do proteins act as command-and-control centers for the activity of our 30,000 genes? And how do they fuel the spread of a pandemic?
The answer is that form follows function: A protein’s shape, or three-dimensional structure, is critical to its biological job. Proteins consist of strings of connected amino acids that fold into very specific shapes—and improper folding can be disastrous. In sickle cell disease, for example, one wrong amino acid in the oxygen-carrying protein hemoglobin causes misfolded protein clumps to stiffen red blood cells. This prevents these normally flexible cells from gliding through the body’s blood vessels: One seemingly small biological error thus starves cells of oxygen and causes the disease’s excruciating pain episodes.
For years, structural biologists like McLellan have analyzed the shapes of proteins in various types of coronaviruses, and this research helped scientists hit the ground running when SARS-CoV-2 emerged. For example, knowing at the very beginning of the COVID-19 outbreak in Wuhan that all coronaviruses use their spike proteins to claw into cells and infect them guided researchers to zero in on the spike protein as a key target for vaccines and drugs.
COVID-19 is a zoonotic disease, which means it jumped from animals to humans. Because they can infect more hosts, these diseases can spread more. Past studies have revealed that mutations in the coronavirus spike protein can change how a coronavirus interacts with a particular host like bats and humans. In addition, they have shown that these viruses use other molecular tricks that can help gaining a foothold in a new species, such as tolerating more errors in copying their genetic material (thus creating new mutations), as well as sticking around long enough to figure out how to hoodwink a new host’s cells, known to virologists as “persistence.”
Applying lessons learned
By early February 2020, just over a month into the pandemic, McLellan and his team had transformed the virus’s genetic sequence into a cryo-EM structure and shared this information with scientists worldwide. About a decade earlier, working in the U.S. National Institutes of Health’s Vaccine Research Laboratory, he and his mentor Barney Graham, MD, PhD, had used structural biology techniques to reengineer an ineffective vaccine by adjusting the shape of a single piece of a protein in a lab-manufactured replica of the respiratory syncytial virus, which isn’t a coronavirus but uses a similar strategy to infect cells.
This time around, teaming up again with Graham and Kizzmekia Corbett, PhD, McLellan and his team, including then-postdoctoral researcher Nianshuang Wang, PhD, used the same playbook. They started looking at SARS-CoV-2 based on what they had learned years before about viruses including other coronaviruses—like those that cause the first SARS, which emerged in 2003, and Middle Eastern respiratory syndrome, or MERS, first reported in 2012.
By introducing just two proline amino acids (out of more than 1000 amino acids) at a key site in the SARS-CoV-2 spike protein, Corbett, Wang, and McLellan custom-shaped a version of the spike that was locked into its most infectious, “prefusion” form and thus was the ideal target for a vaccine.
They shared this information with the biotech company Moderna, and by mid-December 2020, only 11 months later, Moderna had received emergency use authorization from the U.S. Food and Drug Administration for a vaccine that showed 94% protection in the 2020 clinical trials. At least four other major vaccines—from Pfizer, Johnson & Johnson, Novavax, and CureVac—are also based on this team’s version of the spike.
Investments over previous decades in structural biology tools in general, and cryo-EM in particular, had prepared McLellan and others for this moment. As a result, the development and testing of these COVID-19 vaccines set speed records for what is normally a painstaking and laborious process that plods along at biology’s measured pace.
How much speedier? According to McLellan, although we are very close, we still don’t have an effective vaccine against respiratory syncytial virus—and that hunt has been going on for more than 50 years.
Seeing the very, very small
Fortunately for humans, pandemics have been relatively rare. In 1918, the so-called Spanish flu raged around the planet and shut down large sectors of the global economy. Although some viruses are household names, like flu and those that cause the common cold, many arrive without warning. Think Ebola, Zika, the first SARS, and MERS. In each of these cases, basic research using powerful instruments to view viruses in molecular detail has helped humanity understand these threats.
The newest coronavirus is only about 100 nanometers in diameter—very, very small (see the “Scale of the Universe” interactive tool). SARS-CoV-2 is smaller even than a wave of light, meaning it can’t be seen with a typical light microscope because of how light bends or diffracts with very small objects, obscuring them from view.”
Seeing such tiny structures requires a different strategy, using a rapid beam of high-energy electrons. The first electron microscope, the instrument that can do this, was invented in 1931 by two Germans—electrical engineer Max Knoll, PhD, and his student and eventual Nobel laureate, Ernst Ruska— who were then studying the behavior of electrons (watch an animation of how electron microscopy works). Within a few years, the Radio Corporation of America, better known as RCA, made a commercially available version of the instrument that could view various types of very small things: carbon black in car tires, pigments in paints and cosmetics, and, in time, biological specimens like thin slices of tissue.
A related idea conceived in parallel in 1912—that a stream of X-rays bouncing off a sample could trace and recapitulate its shape—gave rise to a similar method called X-ray crystallography. This foundational approach enabled English physicist Francis Crick, PhD; English chemist Rosalind Franklin, PhD; and American biologist James Watson, PhD, to see and understand in 1953 how DNA’s double helical shape allows it to be copied. X-ray crystallography also led to antiretroviral therapy for HIV/AIDS in the 1990s and, several years after that, to a cure for hepatitis C. Just a few weeks into the COVID-19 pandemic, several research groups used X-ray crystallography to nail down the structure of a SARS-CoV-2 spike entwined with its human target, the angiotensin-converting enzyme 2 receptor, known as the ACE2 receptor, on the surface of cells.
Resolution revolution
Although the ability of X-ray crystallography to capture a receptor and its target—or various other molecular ensembles—remains an important application of this technology, a new revolution is already upon us, thanks to cryo-EM, which earned its discoverers the 2017 Nobel Prize in Chemistry. The cat-and-mouse nature of technology development and discovery science has propelled cryo-EM from its early theoretical days in the 1960s to the highly practical, transformative technology it is today in 2021.
Cryo-EM involves flash-freezing a biological sample into a very thin slice of glassy ice, using extremely cold liquid ethane, and then bombarding the sample with electrons. Scientists capture cryo-EM snapshots using a special “camera” that detects electrons directly without using film, and then they use sophisticated computer programs that average millions of images to clean up the equivalent of biological smudges. The end result is a highly precise, atom-by-atom, three-dimensional image (watch an animation of cryo-EM).
Cryo-EM has taken biology by storm, since it offers key advantages over X-ray crystallography: Because a sample is frozen so fast, it’s protected from high-voltage electron damage and does not become dried out and misshapen. And perhaps the most game-changing aspects of cryo-EM are that sample amounts can be much smaller—often one one-thousandth of what’s needed for X-ray crystallography—and that although samples need to be in good shape and reasonably pure, they don’t need to be wrangled into a crystal conformation. That step has been a major technical bottleneck, making it difficult to image many proteins of interest to researchers and drug developers.
Cryo-electron tomography (cryo-ET), used by Sai Li, is an especially promising new frontier, says Bridget Carragher, PhD. Similar to a CT scan of the human body, cryo-ET involves tilting a sample as it’s imaged, creating hundreds or even thousands of two-dimensional images that can be combined by a computer into a three-dimensional reconstruction. The procedure’s key strength is being able to see a sample in its natural environment, such as an organelle within a cell.
Carragher and Clinton Potter, PhD, codirect the New York Structural Biology Center’s Simons electron microscopy department; their work is partially funded by the Simons Foundation to push new boundaries in structural biology using machine learning. Getting to the discovery part of powerful methods like cryo-EM and cryo-ET is “exhausting, time-consuming, and boring,” Carragher says. “With machine learning, we can do this a lot faster, better, and cheaper.”
Cures by design
“Making a protein from scratch on a computer to neutralize a virus sounds like science fiction,” says David Baker, PhD, the director of the University of Washington’s Institute for Protein Design. But that’s exactly what he and his lab have done in an effort to thwart COVID-19.
For 20 years, Baker, who is also a Howard Hughes Medical Institute Investigator, has been designing all kinds of proteins. When COVID-19 came on the scene, he rapidly adapted this longstanding focus of his lab to addressing the medical emergency of the moment. However, he’s quick to note that designing proteins is hardly a plug-and-play operation. “Engineering is what you do when you understand how everything works. You have a procedure for making things and then you repeat it,” Baker says. But science is different: “You start with the problem and work backward to find methods to solve it.”
He and his team, which extends far beyond his lab to include scientists all over the world, run a giant scientific club called the Rosetta Commons. It centers around innovative uses of Rosetta, a computer software program that Baker developed in the late 1990s. Just as the famous stone of the same name once helped linguists decipher ancient languages, the Rosetta software decodes protein shapes that have been scripted by our complex genomic language, which scientists still do not fully understand. Basically, Rosetta uses information about a protein’s amino acid sequence to predict all its possible shapes. The computer program breaks the protein into small segments of amino acid sequences, searches for all the different shapes each segment could assume, and then mixes and matches them until it finds a perfect fit.
Over the past few months, Baker has been using Rosetta “in reverse,” he explains, to create tiny, super-sturdy versions of proteins that fit perfectly into the now precisely defined pockets and grooves of the SARS-CoV-2 spike protein. The intent is to design ideal versions of proteins to block SARS-CoV-2’s function, as well as to act as COVID-19 antiviral drugs. The approach is both powerful and efficient, because he can run millions of simulations to find proteins with specific properties.
“The mini-proteins we’ve been able to design stick to the spike protein as tightly as any known antibody, and they’re much, much more stable,” explains Ian Haydon, who works in Baker’s lab. “You can put them in boiling water and they still work as well as an antibody would.”
Baker predicts that the methods he’s developing now will be extremely powerful for other applications, too—including the next pandemic—and will be even faster. “The next task is to figure out how to do everything we did much more quickly and accurately,” he says. “Rather than taking three or four months to develop a very potent antiviral drug, let’s do it in a week or two after the genomic sequence is released.”
Baker’s work, which is partially funded as a “Big Idea” through the Audacious Project, is heavily focused on tools, the critical enablers of biomedical research. Private philanthropy continues to be an important source of support to keep such discoveries coming.
We don’t know what we don’t know
There are likely countless biological problems in which knowledge lags behind technology—and thus we don’t know what we don’t know until a new tool opens the right window to look at the question.
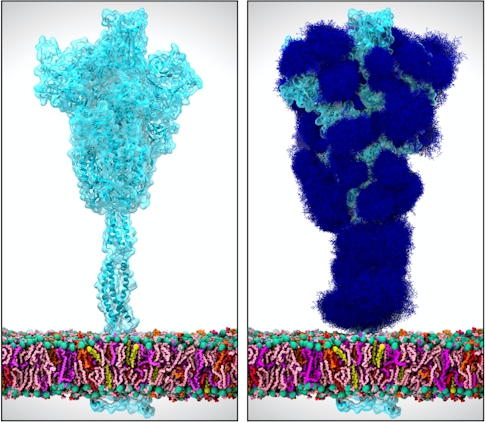
“It’s so powerful when you can see how something works—seeing something completely changes your understanding,” says Rommie Amaro, PhD, of the University of California, San Diego, who uses supercomputers to create simulations of viruses like HIV, influenza, and, most recently, SARS-CoV-2.
Amaro’s “computational microscope” uses supercomputers to combine massive amounts of data about the biological and chemical properties of an organism (or a virus) into a single model of a system: in this case the half-million–atom SARS-CoV-2 particle. Her 28-member international team built a highly realistic model of the SARS-CoV-2 virus that includes its slick lipid coating; its RNA code; its renowned spike proteins; and, in a new wrinkle, the sugary invisibility cloaks worn by the spikes to conceal them from the immune system.
Discovering those sugar cloaks was both surprising and medically important, Amaro explains. “Knowing the unprotected places is critical information for researchers working on COVID-19 drugs,” she says. “These are the vulnerable spots where antibodies, vaccines, and new kinds of drugs can attach.”
Onward after COVID-19
Are we ready for SARS-CoV-3? Or another virus like it?
“I strongly believe it’s important to have interdisciplinary efforts, especially for new technology development,” notes Carragher. “Technology is where many of the biggest breakthroughs come from.”
But conducting ongoing viral imaging work using cutting-edge technologies like cryo-EM and cryo-ET—as well as continually improving them—is expensive and not equitably distributed among scientists who want to use these miracle instruments. And funding for such work doesn’t come easy, Carragher adds. “You have to scrounge it up. . . . We’re very, very fortunate to have Simons Foundation support, but many people don’t.”
The structural biology tools available right now will let scientists hit the ground running to help halt the next invisible monster. But even so, researchers must continue envisioning and perfecting additional revolutionary techniques to reveal still more about SARS-CoV-2—and also about future viruses.
If we’re to be ready for the next pandemic, such vital work must go on.
About the Author
Alison F. Davis is a freelance science writer with a range of clients in biomedicine, including the National Institutes of Health, and a speechwriter and scientific writer for senior scientists and physicians at NIH and elsewhere.
This article first appeared as part of the Science Philanthropy Alliance‘s COVID-19 Basic Science Prequels series. Decades before COVID-19 gripped the world, basic science researchers were laying the groundwork for lifesaving medical and technological breakthroughs now central to the world’s pandemic response. With support from the Kavli Foundation and the Simons Foundation, the Science Philanthropy Alliance has enlisted a team of science writers to explore these science origin stories. Each month, the COVID-19 Basic Science Prequels will unpack the people, history, and serendipitous discovery behind topics that now dominate our daily lives.