Are Similar Processes at Work in Both Brain Development and Cognitive Decline?
On a wintry Tuesday morning during the pandemic, Frederick Bennett, psychiatrist and neuroscience researcher at the University of Pennsylvania, cuddles his 8-week-old infant as he explains how brain cells called microglia develop in mice. Bennett’s daughter has barely begun to beam her first social smiles, but her brain has been firing at a frenetic pace as she learns and grows.
Microglia tend to her newly grown circuits, clearing cellular debris, guarding against tissue damage, and severing unwanted synapses. This constant gardening is critical early in life. Until approximately age 20 for humans, removing synapses is just as crucial to learning as forming them. “I find myself sort of looking at her and thinking about these brain sections of mice at the same time,” Bennett says. “How is all this playing out in her brain?”
Over the last 15 years, scientists have uncovered a wealth of evidence that our innate immune systems and cells within the brain such as microglia, astrocytes and other so-called supporting cells collaborate to prune and maintain connections throughout our lifetimes.
Now, evidence suggests that some of the same processes critical during development also play a central role in normal aging. Signs of cognitive decline, such as forgetting an actor’s name or taking longer to think through a problem, arise not because neurons die but because synapses are lost as we age. Some of the same immune molecules that tag synapses for destruction during development also increase in concentration in the aging brain, suggesting that a similar but faulty pruning process might be at play in cognitive decline. By studying how gene expression changes in microglia over time, scientists have discovered that these cells reactivate certain immune pathways as they age, suggesting parallels between youth and old age.
The research is part of a broader effort to understand the molecular changes that underlie brain aging, which may in turn reveal ways to slow or reverse them. Aging is often portrayed as a kind of unavoidable decay, rather than a condition in and of itself — a perception that needs to change, says Bérénice Benayoun, a gerontology researcher at the University of Southern California. Clues from the immune system may provide key insights. “Among vertebrates, immune dysfunction seems to be one of the key conserved aspects of aging,” says Benayoun.
Understanding these changes could have a major impact on a plethora of diseases. The greatest risk factor for nearly all chronic diseases, including Alzheimer’s disease, cancers and diabetes, is age itself. “Ultimately, if we understand aging, we could fix all of these things in one fell swoop instead of continuing to play whack-a-mole with every disease separately,” says Benayoun, who is an investigator with the Simons Collaboration on Plasticity and the Aging Brain (SCPAB). “We’re treating symptoms of aging rather than the root cause.”
Together for life
Beth Stevens, a neurobiologist at Boston Children’s Hospital and the Broad Institute, began studying how the immune system shapes the brain as a postdoctoral researcher at Stanford University more than a decade ago. Stevens and her colleagues in Ben Barres’ laboratory at Stanford discovered that the developing brain produces immune proteins called complement, which are secreted outside of cells. These proteins accumulate at immature neuronal synapses in the visual system and tag them for elimination. Stevens and colleagues’ study was the first to demonstrate that classical complement proteins, whose role is well studied in the immune system, help direct synaptic pruning during development.
That discovery sparked a keen interest in glial cells and immune proteins. “Beth was probably the first person to put [these immune proteins] on the map [in the context of synaptic pruning],” says George Mentis, a neurobiologist at Columbia University who collaborated with Barres on studies of complement proteins in the brain. Previous research on these cells had focused largely on their role in responding to injury, Alzheimer’s disease and other neurodegenerative conditions.
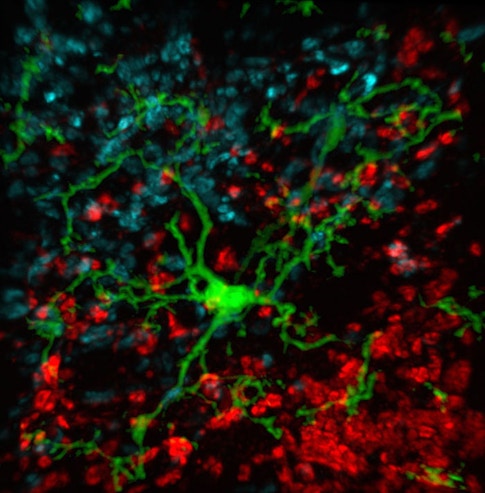
Others in the Barres lab were making similar discoveries about glia and other non-neural cells in the brain. Stevens, an investigator with the SCPAB, recalls watching movies from her colleague Axel Nimmerjahn that showed the dynamic fingerlike projections from microglial cells dancing across neurons and synapses, probing and withdrawing, testing the brain’s delicate connections for flaws to be fixed. These observations, paired with their discovery that complement proteins mark synapses for elimination, suggested that a specific complement protein called C1q may be an instructive ‘eat me’ signal for microglia. The process mirrors immune function outside the brain, where complement proteins tag pathogens for elimination by macrophages. Indeed, follow-up studies by Stevens’ lab and others have provided evidence for this mechanism in both healthy mice and models of age-related disease.
Microglia share some similarities with their non-neural brethren, but also key differences. Just as macrophages clear trash from the lungs, livers and other organs, microglia munch on synapses marked for removal. However, microglia and immune cells that reside in other parts of the body have very different life spans. Immune cells in the blood are constantly refreshed, but microglia are exceptionally long-lived — studies suggest that the microglia present at birth last throughout an animal’s life.
These differences seem to originate very early in development. In mice, microglia develop from progenitor cells that emerge when the embryo is approximately a week old. Those cells grow into so-called primitive macrophages cells and travel to the tissues that will form the fetus’ head. The primitive immune cells develop alongside the fetal brain and are eventually sealed off behind the blood-brain barrier. “Microglia are by far the most fascinating tissue macrophages,” Bennett says. “They’re derived from this early embryonic lineage, and kind of live a parallel life to the rest of the immune system.”
In 2018, Bennett and colleagues showed that microglia’s embryonic origins and tissue environments in the brain govern how the cells develop and function throughout our life span. They engineered mice to lack microglial cells in adulthood, creating a sort of ‘empty canvas,’ and then tried to replace the lost cells by injecting different immune cell precursors into the animals’ brains. Blood-derived stem cells that can form macrophages in other tissues were unable to become true microglia. “They got close, but not all the way there,” Bennett says. “That tells us there’s something special that makes microglia derived from embryonic cells distinct from other macrophages.”
A complex decline
That special status, and their long life spans, could mean that microglia weather life changes differently from other immune cells. Understanding how these cells age could have important implications for understanding — and perhaps slowing — brain aging.
To understand microglia’s role in aging, Stevens and her collaborator Steven McCarroll of Harvard University, also an SCPAB member, used single-cell RNA sequencing to analyze gene expression in individual microglia in mice across their life spans. Early in development, explains Stevens, microglial populations expressed a diverse array of the genes, likely so they could colonize the developing brain, adapt and sculpt synapses early in life. By a month old, most microglia expressed more or less the same genes. But as the cells aged, the researchers noticed that they grouped into smaller subsets, each expressing a signature cluster of genes enriched in distinct molecular signals related to cytokine production and other inflammatory cues. While earlier studies had identified similar changes with age using bulk RNA sequencing, this single-cell approach developed by McCarroll and colleagues enables scientists to probe where these clusters are present in the brain, and how their transcriptional signatures, or ‘states,’ might relate to different functions.
In one recent study, Stevens and her colleagues examined 76,000 individual microglia in young mice, old animals and after a brain injury, and observed changes over time in all three groups. They found nine distinct microglial states, identified by unique patterns of gene expression. Cells from younger animals showed the greatest diversity of genes expressed. Both injured and aged animals had more microglia in an inflammatory state, with distinct subgroups of cells that expressed immune-related genes.
These age-related changes may result in faulty synaptic pruning, albeit not enough to cause problems. Stevens theorizes that two hits to the brain’s microglia — such as aging and brain injury combined — could trigger enough faulty pruning to elicit cognitive decline.
Older microglia seem to struggle with other tasks as well. In 2016, neuroscientist Mikael Simons at the Technical University of Munich and his colleagues found that as mice grew older, the fatty sheath known as myelin that protects their nerve fibers sloughed off more easily. Microglia usually clear this debris, but they were slower to do so in aging brains. In 2-year-old mice (roughly the equivalent of a person in their sixties), chunks of this debris accumulated in microglia, overloading them and making it tougher for them to recover from continued damage to their nerve fibers. These problems are heightened in conditions such as Alzheimer’s disease.
Scientists don’t yet know whether these changes in gene expression and function are an intrinsic trait of aging microglia, or the cells’ response to their aging environments. As sentinel immune cells, microglia may simply be reacting to environmental cues. “If the brain environment changes with age, the microglia are going to respond to that,” Stevens says. “The challenge that lies ahead is to understand the functional consequences of several of these age-related states or populations, such as the chemokine-secreting microglia, on brain plasticity and specific age-related cognitive processes and behaviors.”
One way to assess how the cells’ environment influences their function would be to place younger microglia in aging brains or vice versa. Would swapping younger microglia in an older brain improve trash clearing and other duties and rejuvenate the brain? It’s an idea Bennett plans to test as his research resumes after the pandemic.
However, transplanting cells in the brain is challenging, so neurobiologist Katrin Andreasson at Stanford University and her colleagues took another tack. In January, they reported that even rejuvenating peripheral blood gave aging microglia a boost. When they blocked an inflammatory signal known as PGE2 in the bloodstream of aged mice, microglia reduced their expression of certain pro-inflammatory genes and their production of reactive oxygen species, which exacerbate cellular stress and aging. The animals’ spatial memory and other cognitive functions were identical to those of younger mice. Simply improving peripheral immune function boosted brain function as well, suggesting that the environment is responsible, at least in part, for faulty microglia in aging. (For more on the role of blood factors in aging, see ‘Searching for the Secret of How Young Blood Rejuvenates the Brain.’)
A new view of aging
Increased inflammation, one of the hallmarks of aging, is seen across many tissues and organs in lab-reared animals. One key player appears to be the complement protein C1q, a part of the innate immune system, which Stevens and Barres identified as important for synaptic pruning in young brains. In 2013, Barres and his colleagues showed that in both humans and mice, levels of C1q increased by nearly 300-fold over the course of normal aging. Aged mice deficient in this particular protein showed fewer signs of cognitive and memory decline than those littermates that had typical C1q levels.
Other complement proteins can cause a loss of synapses too. In 2016, McCarroll and his colleagues reported that one such protein, named C4, was overexpressed in people with schizophrenia and piled up at synapses in a mouse model of schizophrenia, causing the animals’ brains to eliminate these tagged connections. Complement proteins are extremely adaptable molecules, McCarroll says. He likens them to molecular Post-it notes: The notes may bear a new message or obscure an existing signal. They may modulate the function of whatever they’re posted on, such as a synapse. Other cells, such as microglia, might ‘read’ and respond to the message. “Systems of molecular Post-it notes evolved to do one thing, but it’s perhaps not hard to adapt them to other purposes,” McCarroll says. “They may well have many different kinds of roles.”
Recognizing that synapse loss and age-dependent cognitive decline occur at around the same time, Stevens and others are now exploring whether the mechanisms of complement-mediated synapse trimming by microglia — the same mechanisms they spotted in early development — are also at work in the aging brain. Stevens says these experiments employ a panoply of recently developed tools, allowing researchers to conditionally alter genes in brain-resident microglia, for example. “We’re now in a position where we can begin to manipulate pathways or specific cell populations that seem important in the single-cell data, and ask what aspects of brain function they mediate,” she says. “We’re just beginning to understand the brain’s immune system and its roles.”
Benayoun and others are exploring why complement protein and other hallmarks of inflammation increase with age even in the absence of infections. One source, she says, are increasing numbers of senescent cells that spew inflammation-triggering chemicals. Another possibility is the gut microbiome. When we’re young, immune cells encounter gut bacteria but their interactions remain confined to intestinal tissues. As we age, this barrier may grow porous, allowing stray bits of bacterial proteins to slip into circulation, where they can spark an inflammatory response. Yet another hypothesis is that virus-derived pieces of DNA that dwell in our genomes are reactivated over time. Immune cells perceive this as a viral infection and launch an inflammatory defense.
“It looks like a lot of mechanisms converge and push the immune system to dysfunction,” Benayoun says. “The problems with sustained neuro-inflammation are that it can lead to inaccurate synapse pruning or cellular damage, or the release of cytokines that damage the blood-brain barrier.”
Benayoun, Stevens and others now plan to conduct fine-scale experiments in different species to understand the interplay of immune changes, age and supporting cells in the brain.
Since these aspects can converge in a multitude of ways to trigger age-related decline, unbiased approaches that can identify all their intersections are crucial, Benayoun says. Some of their tools include genomic and single-cell RNA sequencing, mutant mouse strains, commercially available chemicals that can ablate microglia and new model animals to study aging. “The beauty of these tools is you take a step back and look at everything at once and see how it correlates to function, so we’re not overlooking anything,” she says.
As part of the SCPAB projects, the researchers will look at how gene expression in individual cells changes with age, so as to understand the molecular predictors of brain function changes. “Even if we’re especially interested in microglia, we’re looking at every cell,” Benayoun says. “If the data tell us other cells are more predictive, we’ll still be able to see that.”


