How Clumps of Misshapen Proteins Gum Up Aging Brains
Sometimes, when he gives talks on aging and the human brain, Rick Morimoto jokingly starts with a disclaimer to his audience, to reassure them that what he is about to report need not apply to their own cognitive health. But as he begins describing his research, “the audience, sometimes they get a little nervous,” he says, semi-seriously. The reason? A particularly anxiety-inducing finding from his lab based on a 2014 analysis of autopsied human brains, both young and old. In that research, the team saw that the genetic activity responsible for maintaining the healthy working order of proteins in brain cells drops precipitously with age. “It was frightening,” says Morimoto, a molecular biologist at Northwestern University in Evanston, Illinois. “There was a striking and dramatic decline.”
One consequence of this decline is that otherwise functional proteins can start clumping or aggregating even in normal brains. It’s well known that many neurodegenerative diseases — including Alzheimer’s, Parkinson’s and Huntington’s — are linked to such protein aggregates. These aggregates, collectively called amyloids, are agglomerations of misfolded proteins in the brain. They disrupt neurons and even kill them. But the biggest risk factor for both protein aggregation and neurodegenerative diseases is aging, says the molecular biologist Anne Brunet of Stanford University. “Aging dwarfs all the other factors,” she says.
Indeed, the work of Morimoto and others shows that protein aggregation, besides being a precursor to neurodegenerative disease, may also underlie the normal age-related deterioration of the brain’s ability to learn, remember and process information.
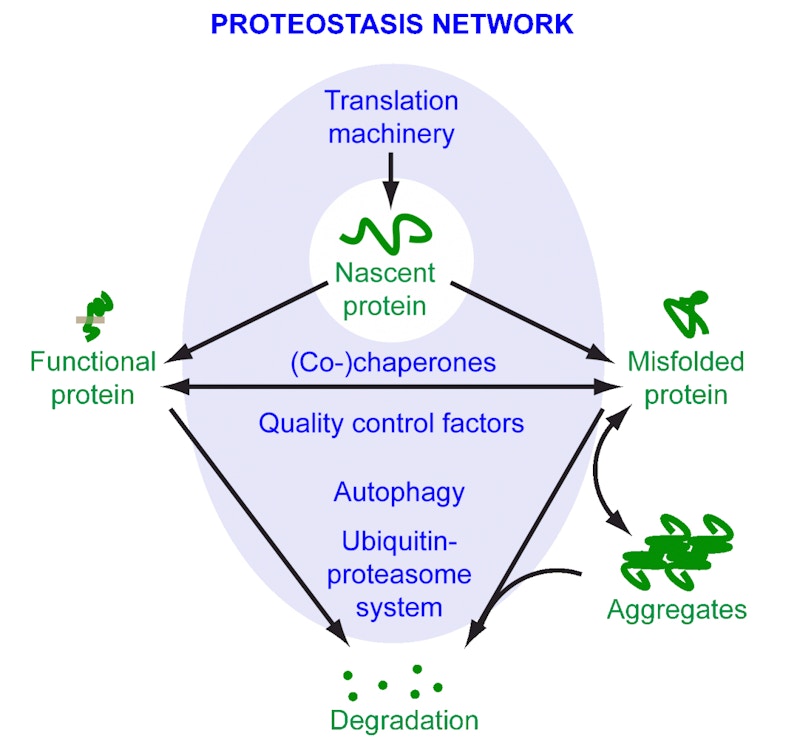
The culprit may be a complex cellular process called proteostasis, which keeps everything in working order. It ensures that the entire set of proteins the organism expresses, known as the proteome, is healthy. This task involves everything from helping proteins to fold correctly to tagging aberrant proteins for degradation and removal. When the process, which is itself accomplished by an intricate network of proteins, is disturbed, misshapen proteins start aggregating into larger clumps. This makes proteostasis a hot topic of study among researchers looking to develop therapies that target protein aggregation.
“If we identify better what happens with the combo of age and those disease aggregates, there would be new ways of slowing or maybe even reversing some aspects of aggregate-based, age-dependent diseases like Alzheimer’s,” says Brunet. Could such therapies also arrest or even reverse the cognitive decline potentially caused by aggregating proteins in the brain? “Yes, it’s possible,” she says.
To this end, researchers are using animal studies to understand the mechanisms behind aging and aggregation. Within the last decade, scientists studying both worms and mice found that protein aggregation increases as the animal ages. Others have linked age-related protein aggregation in rats to cognitive decline. Recent studies have suggested, tentatively, that this could be true in humans too.
The research is now at a critical juncture. Identifying the mechanisms that lead to a breakdown of proteostasis could one day help contain or even reverse the damage. “We’re trying to figure out the mechanism for humans right now. What’s exciting is once you get to mechanism, then I think about it as a switch,” says Morimoto. “If you know what the switch is, you can turn it back on.”
The first signs of aggregation
Morimoto has been at the forefront of such efforts. In 2009, he showed in a landmark experiment that proteostasis collapses with age in the roundworm Caenorhabditis elegans. This is partly due to a reduction in the activity of proteins called chaperones, which make sure that other proteins are folded correctly and are present in the right places inside a cell. Newly synthesized proteins in particular are prone to misfolding and aggregation, so chaperones are at the ready, helping the proteins fold into their functional 3D shapes. Chaperones remain vigilant throughout the life cycle of proteins and are thus an important part of proteostasis. One outcome of the collapse of proteostasis is the accumulation of aggregated proteins, in the brain and elsewhere in the body. “Aggregation is a symptom of cumulative failure,” says Morimoto.
Around the same time, in 2010, the biochemist Della David, who was a postdoc in Cynthia Kenyon’s lab at the University of California, San Francisco, wondered whether proteins other than those implicated in neurodegenerative diseases could also aggregate. The question hadn’t been thoroughly investigated.
In a series of experiments, David, Kenyon and colleagues directly identified hundreds of detergent-insoluble proteins — an indicator of aggregation — in the brains of aging C. elegans worms, the first such finding in a multicellular organism. They also found that delaying aging in genetically manipulated worms also delayed aggregation.
Even as researchers were elucidating the role of aggregated proteins in aging worms, the neurobiologist Carsten Korth of Heinrich Heine University Düsseldorf in Germany and colleagues were looking at mammals, specifically rats. Crucially, they were looking at protein aggregation in the brain. In their work, they found clear indications that proteostasis becomes impaired with age — and that this impairment can be linked to behaviors associated with cognitive decline. Korth’s study involved training rats, both young and old, to find a submerged platform in a pool of water, a standard behavioral task known as the Morris water maze. The worst-performing old rats were considered emblematic of rats with age-associated cognitive decline. The question for the researchers, says Korth, was: “Can we correlate this cognitive difference with proteins that are insoluble only in those specific rats and not in the others?”
In a 2013 publication, Korth and his team showed that they indeed could. They found three proteins, previously implicated in synaptic plasticity, whose insoluble fractions were different in the best-performing older rats compared to the worst-performing older rats. This points to changes in proteostasis as the rats aged. “I believe that proteostasis cannot be maintained as efficiently in aging,” says Korth. “That leads to the diseases, but then also probably leads to the normal cognitive decline during aging.”
Such findings in rats are one thing; discovering the same in humans is entirely another. A hint came courtesy of an analysis of autopsied young and old human brains. In 2014, Morimoto and his team examined levels of gene expression in the brains of neurologically normal accident victims ranging from 20 to 99 years of age. They wondered whether this dataset, maintained by Bruce Yankner’s team at Harvard Medical School, would reveal any changes in the activity of chaperones, those key proteins that help keep proteostasis humming along.
After looking at the expression of about 300 chaperone genes in the individuals’ superior frontal gyrus, they found that about one-third of these genes were repressed with age, whereas one-fifth were more active. Further experiments with C. elegans and human cells in vitro revealed a small subset of these genes that are conserved across worms and humans; in C. elegans, these chaperone proteins have been shown to reduce the toxicity of protein aggregates associated with neurodegenerative diseases. “Sadly, the ones that we identified [as] essential to prevent aggregation all declined in aging [humans], which was sort of like really wonderful bad news,” says Morimoto.
Underactive chaperones are not the only indicator of disturbed proteostasis. Two other major systems, each acting as a sort of cellular clean-up crew, help keep the organisms’ proteome healthy.
The first, called the ubiquitin-proteasome system, tags misshapen or misfolded proteins. These tagged proteins are then targeted for destruction by proteasomes, large protein complexes that break down and flush out aberrant proteins. The second, called autophagy, uses lysosomes to engulf and break down unwanted protein aggregates. Any disruptions to these systems spell trouble for a cell’s proteome.
Recent research has also shown proteasomes and lysosomes to be particularly active in stem cells. “A stem cell has a very unique ability to deal with protein aggregates that no other cell has, because it can’t accumulate this kind of damage,” says Andrew Dillin of the University of California, Berkeley. Otherwise, “it will take itself out, or it will destroy the lineage that it is producing.”
This has opened an entirely new avenue of inquiry when it comes to protein aggregation. By learning how stem cells clear aggregates, scientists could induce these mechanisms in other cells in the body, making them more effective at clearing out the garbage.
Recent findings from Dillin and colleagues provide striking evidence to support this notion. In 2012, Dillin and David Vilchez, who were then colleagues at the Salk Institute for Biological Studies in La Jolla, California, showed that human embryonic stem cells (ESCs) have elevated levels of proteasome activity. This activity decreases in neural progenitor cells (NPCs) derived from the ESCs and diminishes even further as the NPCs differentiate into neurons. This suggests that upregulating proteasomes in neurons could prevent protein aggregation.
Though this approach is promising, some studies have revealed unintended side effects: tumors. “The problem is that when you have that much proteasome activity, it induces cancer [in somatic cells],” says Dillin.
Despite this roadblock, Dillin’s work inspired Brunet and fellow Stanford molecular biologist Judith Frydman and colleagues to better understand how proteostasis deteriorates with age in neural stem cells, and what cognitive deficits arise as a result. Though the work is still in early stages, initial findings suggest ways in which aging neural stem cells could be reinvigorated safely to slow, halt or even reverse cognitive decline.
The research involves quiescent NSCs, a type of neural stem cell that ultimately gives rise to neurons and other brain cells required for olfaction, learning and memory, while also performing repairs following brain injury. Quiescent NSCs can generate activated NSCs, but this process becomes less effective with age — and may help to explain why smell, for example, tends to decline in older adults.
In a series of experiments in mice, the team found that the young, quiescent NSCs contained large lysosomes filled with protein aggregates. By contrast, active NSCs showed greater proteasome activity, suggesting different mechanisms at work for clearing protein aggregates in each NSC type. “We think [the lysosomes] are doing their job in the young animal,” says Brunet. “You also see them in old animals. They are still big. But those lysosomes are less functional. They’re less acidic, so they are less able to degrade protein aggregates.”
These results suggest that reduced lysosome activity may underlie the decline in efficacy of quiescent NSCs during aging. However, when the researchers overexpressed TFEB, a transcription factor thought to regulate lysosomes, they observed a boost in lysosomal activity in the aged quiescent NSCs. “They had fewer aggregates, and they were also better able to activate and proliferate, like stem cells are supposed to do,” says Brunet. The team speculates that the finding may lead to strategies for activating aging neural stem cells in adult animals, which would induce the formation of new neurons.
A new angle of study
To speed up studies of age-related aggregation and its impact on the brain and cognition, Brunet is now turning her attention to a new animal model: the African turquoise killifish. Found in Mozambique and Zimbabwe, these fish reach sexual maturity in just a month and age within four to six months — six times faster than a mouse — making them an extremely viable model for studies of aging. And because the African killifish is a vertebrate, it has organs, a complex brain, an adaptive immune system — and stem cells, says Brunet. Her team is now studying the changes in protein aggregation in the killifish brain and comparing them to similar changes in other organs.
Other work in killifish is already providing promising insights. Alessandro Ori of the Leibniz Institute on Aging-Fritz Lipmann Institute in Jena, Germany, and colleagues recently found that aging killifish brains had greater amount of protein aggregates. In particular, these aggregates contained proteins from ribosomes, part of the protein-making machinery of cells, suggesting that this machinery is being impaired. The disruption “is likely to impair ribosome assembly and to create a pool of orphan proteins at risk of aggregation,” the authors write.
The team also showed that proteasome activity declines in aging killifish. The earlier the onset of reduced proteasome activity, the quicker the fish age and die. This downward spiral may rope in other aspects of proteostasis. Ori and colleagues found that the reduction in the activity of proteasomes caused a compensatory uptick in parts of the lysosomal-autophagy mechanism, possibly putting the other pathways for degrading aggregating proteins under stress. “It is tempting to speculate that a progressive decline of proteasome activity might be the trigger for the impairment of lysosomal function that characterizes aging and late‐onset neurodegenerative disorders,” the authors write. “The combination of reduced proteasome activity and impaired lysosome/autophagy would make old brains more vulnerable to the accumulation of protein aggregates, neuronal loss, and consequently, favor the onset of neurodegenerative disorders.”
Whether they focus on worms, rodents or fish, the ultimate goal of all these studies is to apply this knowledge to the human brain. Indeed, a recent study of autopsied human brains showed that individuals diagnosed with Alzheimer’s disease and mild cognitive decline had more protein aggregates when compared to healthy controls, including aggregates of proteins not typically associated with Alzheimer’s. “Our theory was that there’s probably a lot of other proteins aggregating that are probably important,” says Devin Kepchia of the Salk Institute, who led the study.
The team did in fact identify aggregates of many such proteins, both in Alzheimer’s patients and in those with mild cognitive impairment, compared to controls. These included proteins involved in glycolysis (a metabolic process that breaks down glucose) and in the ubiquitin-proteasome pathway, as well as proteins needed for DNA repair. “Previous experiments have shown that if you take these [DNA repair] proteins out of the animal models, if you knock them out of mice, it creates phenotypes of increased aging,” says Kepchia. “So it kind of all falls together with the fact that, with aggregation, we have potentially increased aging rates, we have loss of ability to maintain protein homeostasis, and we also have altered metabolism.”
Taken together, these results strongly suggest that protein aggregates are inherently bad — and that the key to ameliorating the negative effects of aging is to find a safe way to get rid of them. That being said, some scientists, including Dillin, strike a cautionary note. “When people want to start breaking down these aggregates or getting rid of them, I get worried, because then are we going to start creating more toxicity?” says Dillin. Simply clearing aggregates could be akin to treating the symptom and not the cause.
Indeed, at first blush, decades of groundbreaking work — in worms, in rodents, in fish and now in humans — seem to point to the decline of proteostasis as a central cause of protein aggregation. But proteostasis arises from a complex, responsive network of proteins — one that can adapt to stresses when we are young but fails to do so as we age. “That’s what a lot of the proteostasis researchers are trying to figure out. What did fall apart late in life? And can we put it back later on?” says Dillin. “But it’s a network, and just putting one component of the network back is probably not going to be sufficient. You have to restore the entire network.”


